


Pearls from the Pangenome
Progress 23 years after the first human genome draft sequence
A notable series of publications in Nature and Nature Biotechnology this week provide a major expansion of knowledge about the human genome, filling in important gaps that have existed for over 20 years. It’s called the Pangenome.

You may recall where we were in June 2000 with an early first draft (as reported on front page of the New York Times below). That reference genome sequence was predominantly based on one individual from Buffalo, New York. It was full of gaping holes and mistakes. The work of the Human Pangenome Reference Consortium Project, led by University of California, Santa Cruz (Benedict Paten and Karen Miga) with international collaborators, has now provided a new resource, a far more complete and accurate human genome sequence from 47 diverse individuals, with a planned total number of 350 individuals to be completed in 2024. It’s a 5-year initiative that is supported by the National Institutes of Health at a cost of about $40 million.

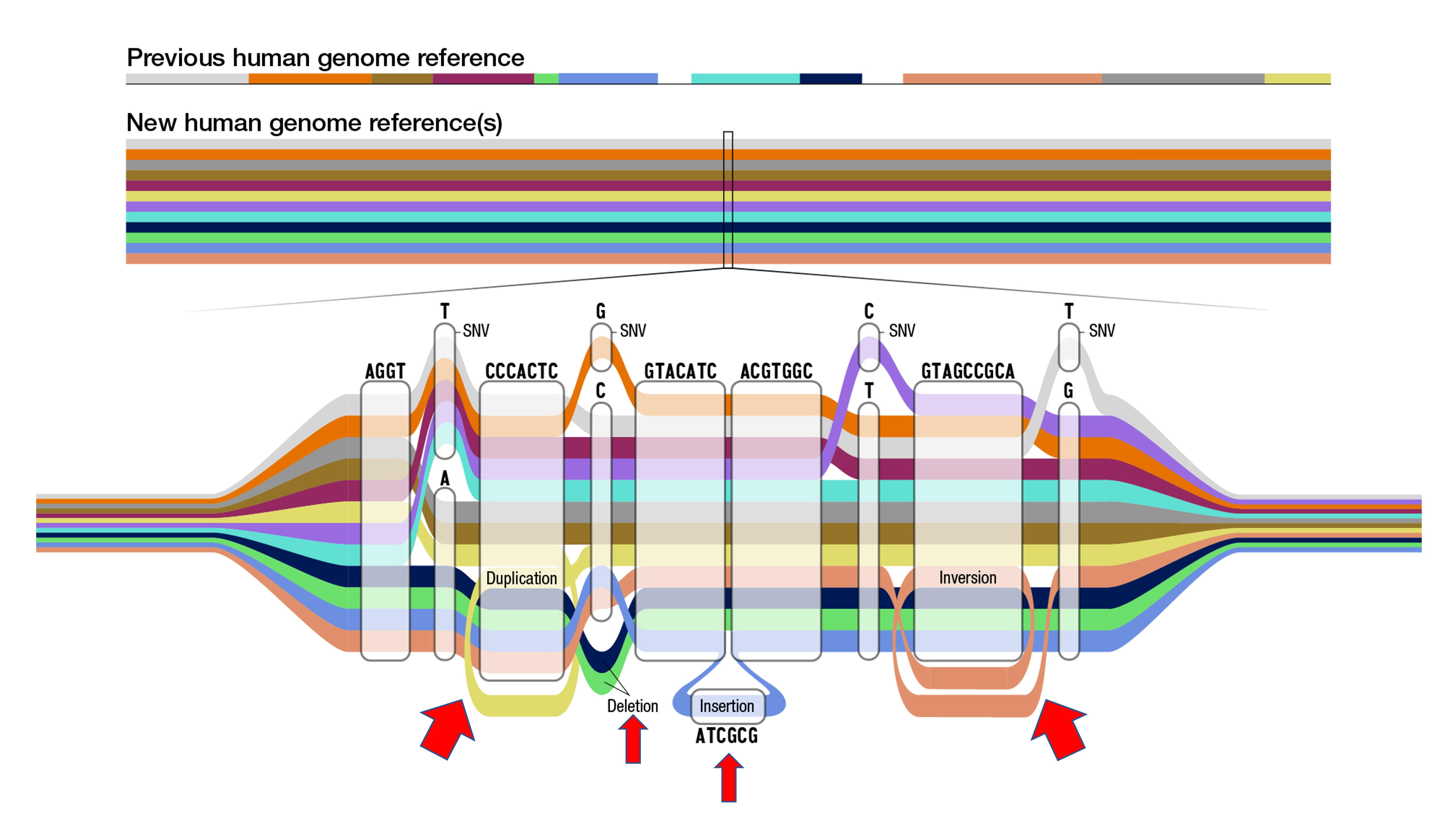
This accomplishment is an outgrowth of exceptionally hard, painstaking work that has relied on next-generation sequencing tools, computational biology and analytics. Back in the early 2000s the variation in the genome that was mapped centered on single nucleotide (or base) letters or polymorphisms (SNVs below, also commonly referred to as SNPs), like an A for a T or a C for a G. The new pangenome has identified nearly 120 million nucleotides to add on to the ~3 billion, and most of these were structural variations which include segmental duplications, deletions, insertions and inversions, as schematically shown below. These structural variants, with 50 or more bases, are notoriously hard to sequence, and 70% had not been adequately characterized (see red arrows below). They account for about 90 of the 120 million new letters. The pangenome as it stands today increases detection of structural variants by more than 100% and of SNVs by 34%—significant jumps in accuracy. Beyond accuracy, the sequence diversity within segmental duplications is noteworthy, with the finding they are 60% more diverse than non-repetitive regions of the genome and exhibit marked inter-individual divergence.
Importantly, the genomes for all 47 diverse individuals are mapped diploid or “phased”—that is the mother and father haplotypes are fully resolved, which actually represents 94 unique complete genome sequences. Each genome was assembled de novo—from scratch. This diploid, de novo feat wasn’t possible 23 years ago and will help our future understanding of how genes (and regions of the genome outside of genes) are inherited.
Overall, the current pangenome is >99% accurate for base pairs and structural variants and >99% complete. The Pangenome is pan-ancestry with African, Asian and European ancestries represented.
Our genomes vary only sightly from one person to another, with 99.6% the same. But the variation of that 0.4% occurs throughout the 3 billion bases, and understanding the full spectrum of variants—which turned out to be far more complex than envisioned in 2000—is pivotal to understanding the links to health and diseases. While many of the variants are “cosmopolitan,” similar across ancestries, a large proportion are ancestry-specific, which underscores how important it is to have a fully diverse reference genome. It’s the differences in the ~0.4% that makes each of us unique.
The new features of defining structural variations and the diploid genome were made possible, at least in part, by long read sequencing with such methodology enabled by Pacific Biosciences and Oxford Nanopore. A key contributor to the progress is related to computational methodology, with advanced algorithms for accurate detection of genome variations.
Another point of progress is moving beyond linear, which until now has largely been the way regions of the genome are represented. Now with so many of the structural variants and recombinations revealed, we’re seeing “cactus” graphs to more accurately portray them, as seen by the “snarls” (also known as bubbles)— the branching and merging paths— in the graph above, and the one below from the main publication.
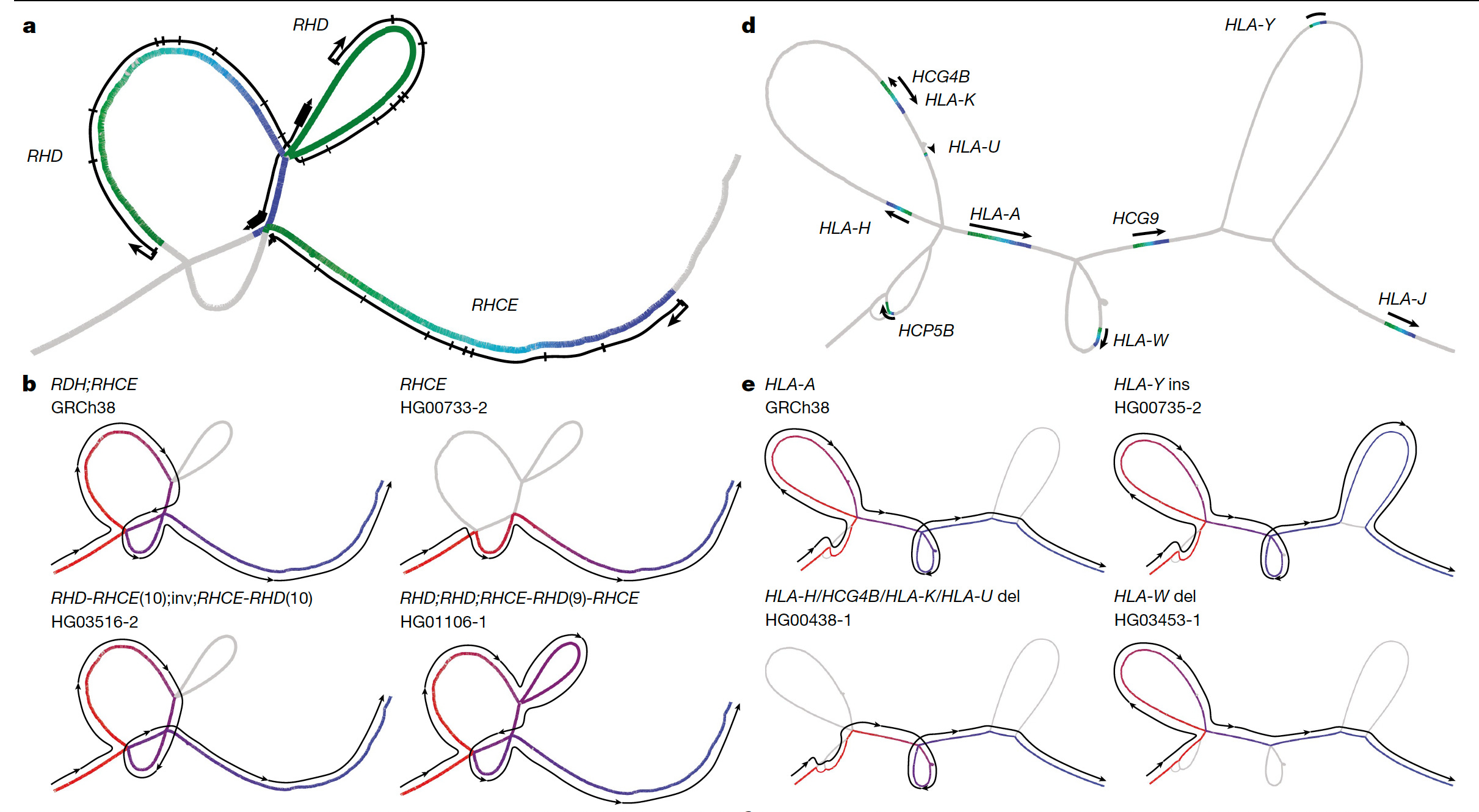
Below is a simplified graphic to show the geographic origin of the 47 individuals with the 94 whole genome sequences (mother and father partitioned) along with the “underground-railway” map with the boxed portions representing sequences found in one or more genomes at that site and the branches showing variation.

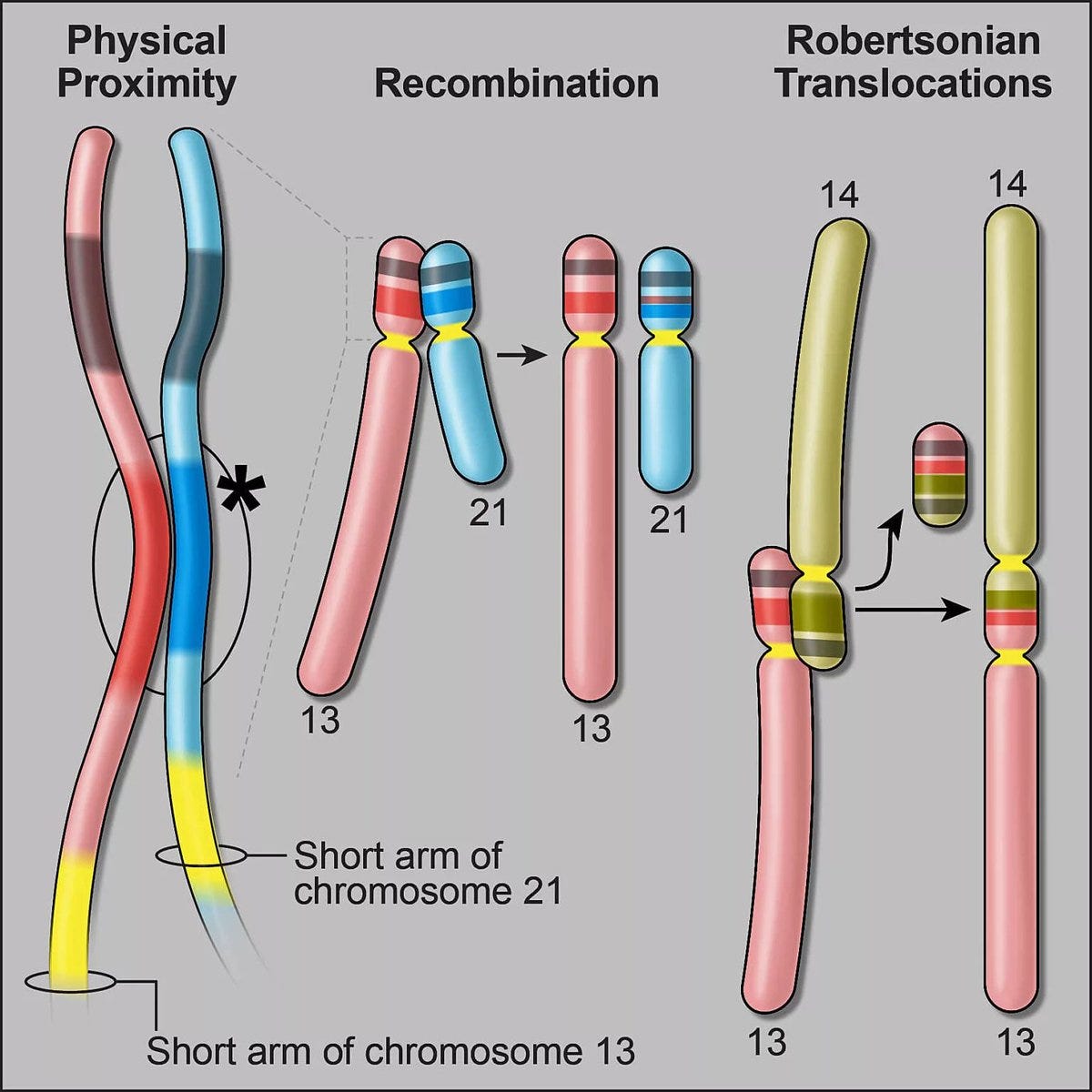
A very interesting discovery related to the chromosomes with a p-arm much shorter than the q-arm, so called acrocentric chromosomes (chromosome 13, 14, 15, 21, and 22). From the pangenome assembly and bioinformatics work, for the first time it was revealed precisely how a piece of one chromosome breaks off and fuses with another to form Robertson translocations, among mismatched chromosome pairs, as seen below, which are not infrequent, occurring in about 1 of 800 births. These recombinations were first observed 50 years ago but the mechanism was finally cracked by this chromosome 14 inverted segmental duplication. Apart from a higher risk of infertility, such translocations are generally not attributable to other medical conditions. But carriers with this form of trisomy (3 copies of a part of a chromosome instead of a pair) are likely more frequent than generally appreciated. It’s just one example of how the pangenome can deconvolute longstanding gaps in our understanding of human genomics.
In sum, to use a metaphor, Ewan Birney (European Molecular Biology Laboratory, EMBL Deputy Director) via a twitter thread, aptly put it “the pangenome is like the complete works of Shakespeare including all the big variations in quarto and folios and anything else. And by having this we can better take any person's DNA and analyse it appropriately.”
The Most Complete Human Genome
Of course, there were many iterations of the reference human genome over the past 2 decades, as exemplified by a series of 6 publications in Science on the more complete genome last year. This was the outgrowth of the Telomere-to-Telomere (T2T) Consortium zooming in on the centromere and telomere parts of the chromosome that were previously indecipherable, but revealed through long-read sequencing methodology and sequencing a cell line from a complete hydatidiform mole (CHM). By removing gaps in about 8% of the genome, 200 million bases were added to the reference sequence (including 3.9 million SNVs), especially segmental duplications, such as found in the short arm of chromosomes, and mobile elements—genomic regions from virus that had incorporated into the genome. It helped provide links of genomic variations to over 600 diseases. This was substantial progress from the latest 2017 reference version known as GRCh38. A good graphic to convey this is from Deanna Church’s accompanying commentary below.

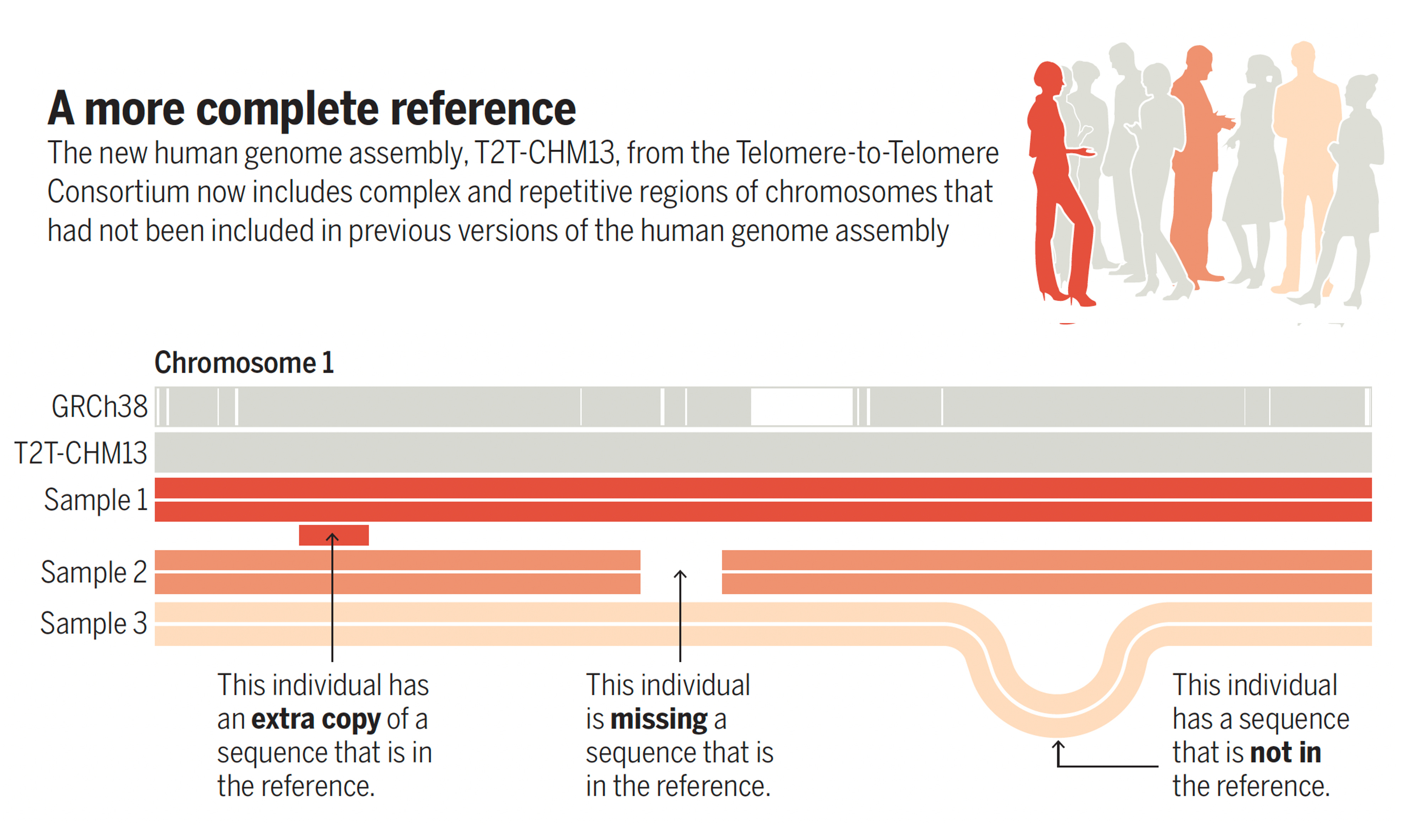
Why is this a big step forward?
The human reference genome is used for countless applications. A front page article in The Washington Post this week highlighted a big one—diagnosis of rare diseases.

An example is Celia Steele, a 10-year-old who has never been able to walk or talk and requires oxygen supplementation and a feeding tube. She has been found to have 2 mutations in the gene PDE2A, 1 derived from each parent as carriers (with no clinical manifestations), and she is one of five people in the world who carries such mutations that have been identified to date. That’s exceptionally rare but the cumulative burden of rare genomic diseases, about 10,000 such diseases in aggregate, affects as many as 30 million Americans. No doubt this revamped reference genome will help unraveling the genomic underpinnings of many rare diseases and people with a serious medical condition of unknown (“idiopathic”) cause.
The other critical advance is diversity of the pangenome. For many years the work in sequencing and genome-wide association studies were predominantly performed in people of European ancestry. It took a long time to get studies at scale which included individuals of African and Asian ancestry. The pangenome published this week is a draft, a work in progress, which will get even deeper and richer with the completion of the 350 individuals by the mid-2024. All of this is open source to researchers, the open science tradition in the human genomics community which has catalyzed much progress being made in the field, even though some of it it may appear to be long overdue.
Regarding the new, enhanced pangenome diversity, Eric Green, director of the National Human Genome Research Institute, said: “There is so much frustration in medicine about how we are operating with such a lack of fundamental knowledge. We will look back on how we practiced medicine, and we will be astonished at how barbaric it was.”
Learning about the importance of diversity for this multi-decade history of human genomics is transferable to all of life science and medical disciplines. In recent months, we’re seeing the intense interest in large language AI models, which carry all the inherent biases of the inputs to the models. An essential learning from human genomics is to squarely address the diversity issue as early as possible; we’ve already seen a multitude of flagrant examples of bias in AI algorithms in the medical space.
The Pangenome is a real ground truth, the reference source for so much work being done in genomic medicine. Even though it’s a fairly complex topic, I couldn’t resist putting it in context for the namesake of this newsletter!
Appreciative of your reading, sharing, and subscribing to Ground Truths.
And much gratitude to the paid subscribers who have helped fund many of our high school and college summer interns at Scripps Research.