


The most enthralling conversation I’ve ever had with anyone on cancer.
It’s with Charlie Swanton who is a senior group leader at the Francis Crick Institute, the Royal Society Napier Professor in Cancer and medical oncologist at University College London, co-director of Cancer Research UK.
Video snippet from our conversation. Full videos of all Ground Truths podcasts can be seen on YouTube here. The audios are also available on Apple and Spotify.
Transcript with audio links and many external links
Eric Topol (00:07):
Well, hello, this is Eric Topol with Ground Truths, and I am really fortunate today to connect us with Charlie Swanton, who is if not the most prolific researcher in the space of oncology and medicine, and he's right up there. Charlie is a physician scientist who is an oncologist at Francis Crick and he heads up the lung cancer area there. So Charlie, welcome.
Charles Swanton (00:40):
Thank you, Eric. Nice to meet you.
Learning from a Failure
Eric Topol (00:43):
Well, it really is a treat because I've been reading your papers and they're diverse. They're not just on cancer. Could be connecting things like air pollution, it could be Covid, it could be AI, all sorts of things. And it's really quite extraordinary. So I thought I'd start out with a really interesting short paper you wrote towards the end of last year to give a sense about you. It was called Turning a failing PhD around. And that's good because it's kind of historical anchoring. Before we get into some of your latest contributions, maybe can you tell us about that story about what you went through with your PhD?

Charles Swanton (01:26):
Yeah, well thank you, Eric. I got into research quite early. I did what you in the US would call the MD PhD program. So in my twenties I started a PhD in a molecular biology lab at what was then called the Imperial Cancer Research Fund, which was the sort of the mecca for DNA tumor viruses, if you like. It was really the place to go if you wanted to study how DNA tumor viruses worked, and many of the components of the cell cycle were discovered there in the 80s and 90s. Of course, Paul Nurse was the director of the institute at the time who discovered cdc2, the archetypal regulator of the cell cycle that led to his Nobel Prize. So it was a very exciting place to work, but my PhD wasn't going terribly well. And sort of 18, 19 months into my PhD, I was summoned for my midterm reports and it was not materializing rapidly enough.
(02:25):
And I sat down with my graduate student supervisors who were very kind, very generous, but basically said, Charlie, this isn't going well, is it? You've got two choices. You can either go back to medical school or change PhD projects. What do you want to do? And I said, well, I can't go back to medical school because I’m now two years behind. So instead I think what I'll do is I'll change PhD projects. And they asked me what I'd like to do. And back then we didn't know how p21, the CDK inhibitor bound to cyclin D, and I said, that's what I want to understand how these proteins interact biochemically. And they said, how are you going to do that? And I said, I'm not too sure, but maybe we'll try yeast two-hybrid screen and a mutagenesis screen. And that didn't work either. And in the end, something remarkable happened.
(03:14):
My PhD boss, Nic Jones, who's a great guy, still is, retired though now, but a phenomenal scientist. He put me in touch with a colleague who actually works next door to me now at the Francis Crick Institute called Neil McDonald, a structural biologist. And they had just solved, well, the community had just solved the structure. Pavletich just solved the structure of cyclin A CDK2. And so, Neil could show me this beautiful image of the crystal structure in 3D of cyclin A, and we could mirror cyclin D onto it and find the surface residue. So I spent the whole of my summer holiday mutating every surface exposed acid on cyclin D to an alanine until I found one that failed to interact with p21, but could still bind the CDK. And that little breakthrough, very little breakthrough led to this discovery that I had where the viral cyclins encoded by Kaposi sarcoma herpes virus, very similar to cyclin D, except in this one region that I had found interactive with a CDK inhibitor protein p21.
(04:17):
And so, I asked my boss, what do you think about the possibility this cyclin could have evolved from cyclin D but now mutated its surface residues in a specific area so that it can't be inhibited by any of the control proteins in the mammalian cell cycle? He said, it's a great idea, Charlie, give it a shot. And it worked. And then six months later, we got a Nature paper. And that for me was like, I cannot tell you how exciting, not the Nature paper so much as the discovery that you were the first person in the world to ever see this beautiful aspect of evolutionary biology at play and how this cyclin had adapted to just drive the cell cycle without being inhibited. For me, just, I mean, it was like a dream come true, and I never experienced anything like it before, and I guess it's sizes the equivalent to me of a class A drug. You get such a buzz out of it and over the years you sort of long for that to happen again. And occasionally it does, and it's just a wonderful profession.
Eric Topol (05:20):
Well, I thought that it was such a great story because here you were about to fail. I mean literally fail, and you really were able to turn it around and it should give hope to everybody working in science out there that they could just be right around the corner from a significant discovery.
Charles Swanton (05:36):
I think what doesn't break you makes you stronger. You just got to plow on if you love it enough, you'll find a way forward eventually, I hope.
Tracing the Evolution of Cancer (TRACERx)
Eric Topol (05:44):
Yeah, no question about that. Now, some of your recent contributions, I mean, it's just amazing to me. I just try to keep up with the literature just keeping up with you.
Charles Swanton (05:58):
Eric, it's sweet of you. The first thing to say is it's not just me. This is a big community of lung cancer researchers we have thanks to Cancer Research UK funded around TRACERx and the lung cancer center. Every one of my papers has three corresponding authors, multiple co-first authors that all contribute in this multidisciplinary team to the sort of series of small incremental discoveries. And it's absolutely not just me. I've got an amazing team of scientists who I work with and learn from, so it's sweet to give me the credit.
Eric Topol (06:30):
I think what you're saying is really important. It is a team, but I think what I see through it all is that you're an inspiration to the team. You pull people together from all over the world on these projects and it's pretty extraordinary, so that's what I would say.
Charles Swanton (06:49):
The lung community, Eric, the lung cancer community is just unbelievably conducive to collaboration and advancing understanding of the disease together. It's just such a privilege to be working in this field. I know that sounds terribly corny, but it is true. I don't think I recall a single email to anybody where I've asked if we can collaborate where they've said, no, everybody wants to help. Everybody wants to work together on this challenge. It's just such an amazing field to be working in.
Eric Topol (07:19):
Yeah. Well I was going to ask you about that. And of course you could have restricted your efforts or focused on different cancers. What made you land in lung cancer? Not that that's only part of what you're working on, but that being the main thing, what drew you to that area?
Charles Swanton (07:39):
So I think the answer to your question is back in 2008 when I was looking for a niche, back then it was lung cancer was just on the brink of becoming an exciting place to work, but back then nobody wanted to work in that field. So there was a chair position in thoracic oncology and precision medicine open at University College London Hospital that had been open, as I understand it for two years. And I don't think anybody had applied. So I applied and because I was the only one, I got it and the rest is history.
(08:16):
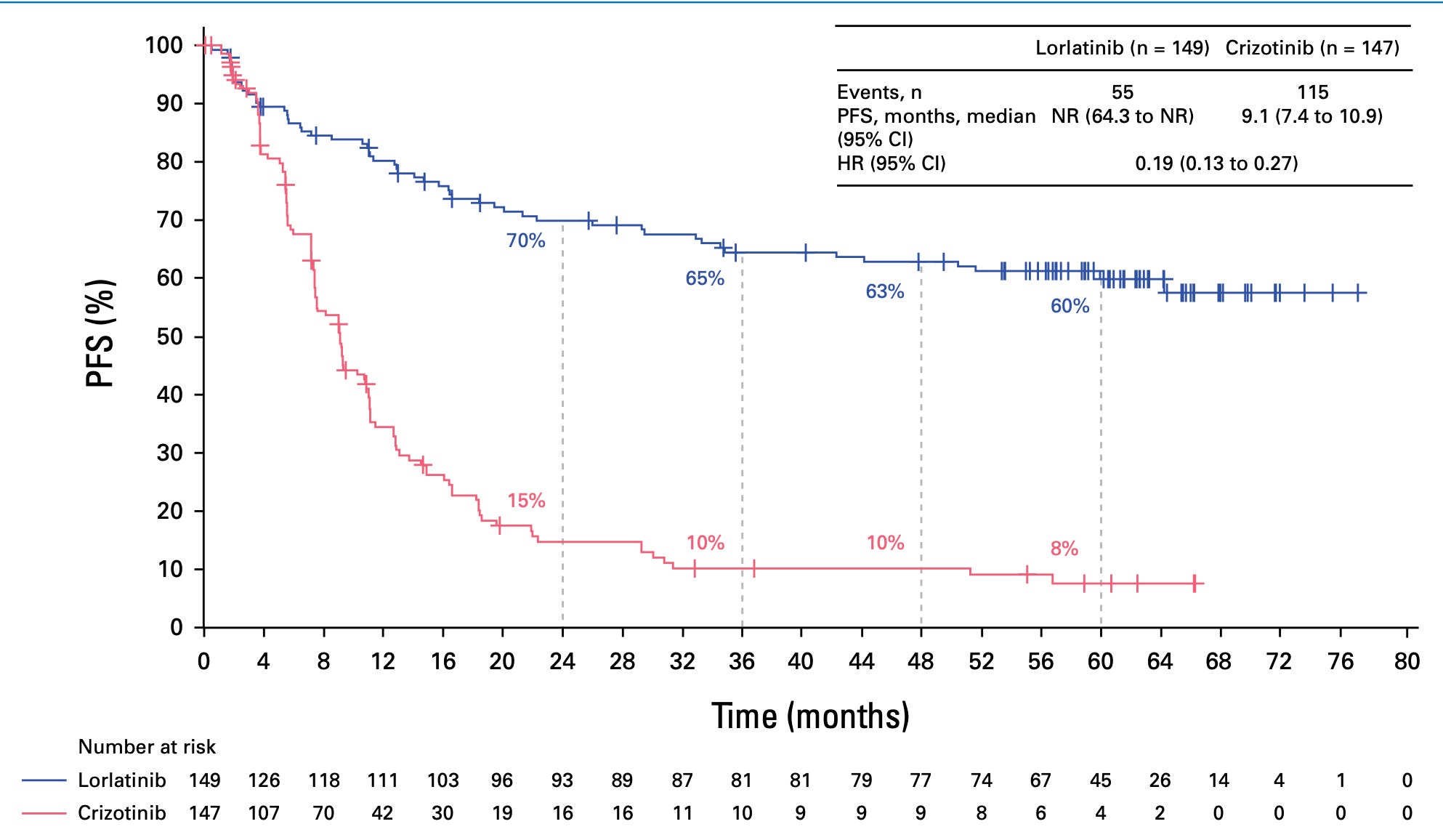
And of course that was right at the time when the IPASS draft from Tony Mok was published and was just a bit after when the poster child of EGFR TKIs and EGFR mutant lung cancer had finally proven that if you segregate that population of patients with EGFR activating mutation, they do incredibly well on an EGFR inhibitor. And that was sort of the solid tumor poster child along with Herceptin of precision medicine, I think. And you saw the data at ASCO this week of Lorlatinib in re-arranged lung cancer. Patients are living way beyond five years now, and people are actually talking about this disease being more like CML. I mean, it's extraordinary the progress that's been made in the last two decades in my short career.
Eric Topol (09:02):
Actually, I do want to have you put that in perspective because it's really important what you just mentioned. I was going to ask you about this ASCO study with the AKT subgroup. So the cancer landscape of the lung has changed so much from what used to be a disease of cigarette smoking to now one of, I guess adenocarcinoma, non-small cell carcinoma, not related to cigarettes. We're going to talk about air pollution in a minute. This group that had, as you say, 60 month, five year plus survival versus what the standard therapy was a year plus is so extraordinary. But is that just a small subgroup within small cell lung cancer?
Charles Swanton (09:48):
Yes, it is, unfortunately. It’s just a small subgroup. In our practice, probably less than 1% of all presentations often in never smokers, often in female, never smokers. So it is still in the UK at least a minority subset of adenocarcinomas, but it's still, as you rightly say, a minority of patients that we can make a big difference to with a drug that's pretty well tolerated, crosses the blood-brain barrier and prevents central nervous system relapse and progression. It really is an extraordinary breakthrough, I think. But that said, we're also seeing advances in smoking associated lung cancer with a high mutational burden with checkpoint inhibitor therapy, particularly in the neoadjuvant setting now prior to surgery. That's really, really impressive indeed. And adjuvant checkpoint inhibitor therapies as well as in the metastatic setting are absolutely improving survival times and outcomes now in a way that we couldn't have dreamt of 15 years ago. We've got much more than just platinum-based chemo is basically the bottom line now.
Revving Up Immunotherapy
Eric Topol (10:56):
Right, right. Well that actually gets a natural question about immunotherapy also is one of the moving parts actually just amazing to me how that's really, it's almost like we're just scratching the surface of immunotherapy now with checkpoint inhibitors because the more we get the immune system revved up, the more we're seeing results, whether it's with vaccines or CAR-T, I mean it seems like we're just at the early stages of getting the immune system where it needs to be to tackle the cancer. What's your thought about that?
Charles Swanton (11:32):
I think you're absolutely right. We are, we're at the beginning of a very long journey thanks to Jim Allison and Honjo. We've got CTLA4 and PD-1/PDL-1 axis to target that's made a dramatic difference across multiple solid tumor types including melanoma and lung cancer. But undoubtedly, there are other targets we've seen LAG-3 and melanoma and then we're seeing new ways, as you rightly put it to mobilize the immune system to target cancers. And that can be done through vaccine based approaches where you stimulate the immune system against the patient's specific mutations in their cancer or adoptive T-cell therapies where you take the T-cells out of the tumor, you prime them against the mutations found in the tumor, you expand them and then give them back to the patient. And colleagues in the US, Steve Rosenberg and John Haanen in the Netherlands have done a remarkable job there in the context of melanoma, we're not a million miles away from European approvals and academic initiated manufacturing of T-cells for patients in national health systems like in the Netherlands.
(12:50):
John Haanen's work is remarkable in that regard. And then there are really spectacular ways of altering T-cells to be able to either migrate to the tumor or to target specific tumor antigens. You mentioned CAR-T cell therapies in the context of acute leukemia, really extraordinary developments there. And myeloma and diffuse large B-cell lymphoma as well as even in solid tumors are showing efficacy. And I really am very excited about the future of what we call biological therapies, be it vaccines, an antibody drug conjugates and T-cell therapies. I think cancer is a constantly adapting evolutionary force to be reckoned with what better system to combat it than our evolving immune system. It strikes me as being a future solution to many of these refractory cancers we still find difficult to treat.
Eric Topol (13:48):
Yeah, your point is an interesting parallel how the SARS-CoV-2 virus is constantly mutating and becoming more evasive as is the tumor in a person and the fact that we can try to amp up the immune system with these various means that you just were reviewing. You mentioned the other category that's very hot right now, which is the antibody drug conjugates. Could you explain a bit about how they work and why you think this is an important part of the future for cancer?
Antibody-Drug Conjugates
Charles Swanton (14:26):
That's a great question. So one of the challenges with chemotherapy, as you know, is the normal tissue toxicity. So for instance, neutropenia, hair loss, bowel dysfunction, diarrhea, epithelial damage, essentially as you know, cytotoxics affect rapidly dividing tissues, so bone marrow, epithelial tissues. And because until relatively recently we had no way of targeting chemotherapy patients experienced side effects associated with them. So over the last decade or so, pioneers in this field have brought together this idea of biological therapies linked with chemotherapy through a biological linker. And so one poster chart of that would be the drug T-DXd, which is essentially Herceptin linked to a chemotherapy drug. And this is just the most extraordinary drug that obviously binds the HER2 receptor, but brings the chemotherapy and proximity of the tumor. The idea being the more drug you can get into the tumor and the less you're releasing into normal tissue, the more on tumor cytotoxicity you'll have and the less off tumor on target normal tissue side effects you'll have. And to a large extent, that's being shown to be the case. That doesn't mean they're completely toxicity free, they're not. And one of the side effects associated with these drugs is pneumonitis.
(16:03):
But that said, the efficacy is simply extraordinary. And for example, we're having to rewrite the rule books if you like, I think. I mean I'm not a breast cancer physician, I used to be a long time ago, but back in the past in the early 2000s, there was HER2 positive breast cancer and that's it. Now they're talking about HER2 low, HER2 ultra-low, all of which seem to in their own way be sensitive to T-DXd, albeit to a lower extent than HER2 positive disease. But the point is that there doesn't seem to be HER2 completely zero tumor group in breast cancer. And even the HER2-0 seem to benefit from T-DXd to an extent. And the question is why? And I think what people are thinking now is it's a combination of very low cell service expression of HER2 that's undetectable by conventional methods like immunohistochemistry, but also something exquisitely specific about the way in which HER2 is mobilized on the membrane and taken back into the cell. That seems to be specific to the breast cancer cell but not normal tissue. So in other words, the antibody drug conjugate binds the tumor cell, it's thought the whole receptor's internalized into the endosome, and that's where the toxicity then happens. And it's something to do with the endosomal trafficking with the low level expression and internalization of the receptor. That may well be the reason why these HER2 low tumors are so sensitive to this beautiful technology.
Eric Topol (17:38):
Now I mean it is an amazing technology in all these years where we just were basically indiscriminately trying to kill cells and hoping that the cancer would succumb. And now you're finding whether you want to call it a carry or vector or Trojan horse, whatever you want to call it, but do you see that analogy of the HER2 receptor that's going to be seen across the board in other cancers?
Charles Swanton (18:02):
That's the big question, Eric. I think, and have we just lucked out with T-DXd, will we find other T-DXd like ADCs targeting other proteins? I mean there are a lot of ADCs being developed against a lot of different cell surface proteins, and I think the jury's still out. I'm confident we will, but we have to bear in mind that biology is a fickle friend and there may be something here related to the internalization of the receptor in breast cancer that makes this disease so exquisitely sensitive. So I think we just don't know yet. I'm reasonably confident that we will find other targets that are as profoundly sensitive as HER2 positive breast cancer, but time will tell.
Cancer, A Systemic Disease
Eric Topol (18:49):
Right. Now along these lines, well the recent paper that you had in Cell, called embracing cancer complexity, which we've talking about a bit, in fact it's kind of those two words go together awfully well, but hallmarks of systemic disease, this was a masterful review, as you say with the team that you led. But can you tell us about what's your main perspective about this systemic disease? I mean obviously there's been the cancer is like cardiovascular and cancers like this or that, but here you really brought it together with systemic illness. What can you say about that?
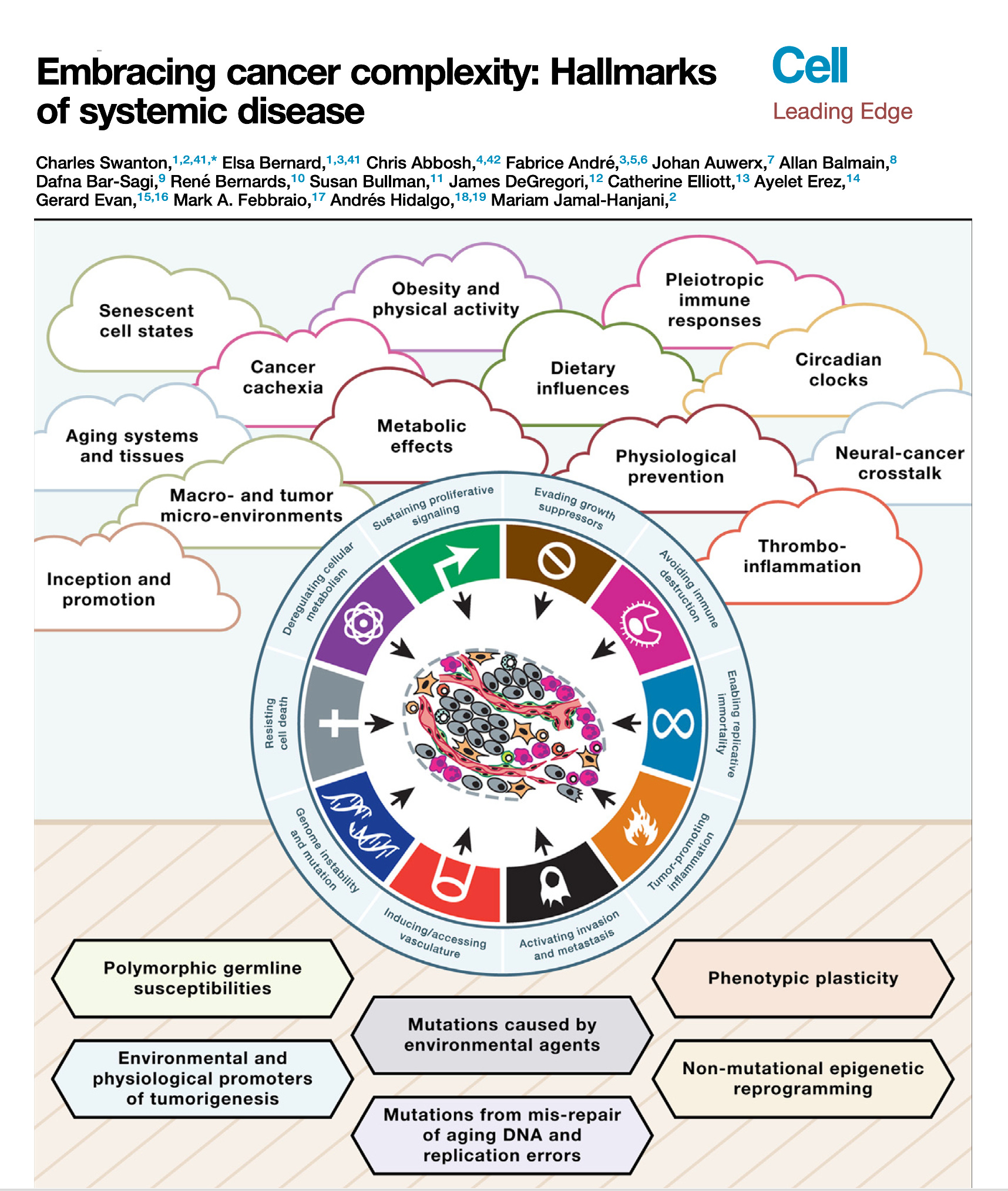
Charles Swanton (19:42):
Well, thanks for the question first of all, Eric. So a lot of this comes from some of my medical experience of treating cancer and thinking to myself over the years, molecular biology has had a major footprint on advances in treating the disease undoubtedly. But there are still aspects of medicine where molecular biology has had very little impact, and often that is in areas of suffering in patients with advanced disease and cancer related to things like cancer cachexia, thrombophilia. What is the reason why patients die blood clots? What is the reason patients die of cancer at all? Even a simple question like that, we don't always know the answer to, on death certificates, we write metastatic disease as a cause of cancer death, but we have patients who die with often limited disease burden and no obvious proximal cause of death sometimes. And that's very perplexing, and we need to understand that process better.
(20:41):
And we need to understand aspects like cancer pain, for example, circadian rhythms affect biological sensitivity of cancer cells to drugs and what have you. Thinking about cancer rather than just sort of a single group of chaotically proliferating cells to a vision of cancer interacting both locally within a microenvironment but more distantly across organs and how organs communicate with the cancer through neuronal networks, for example, I think is going to be the next big challenge by setting the field over the next decade or two. And I think then thinking about more broadly what I mean by embracing complexity, I think some of that relates to the limitations of the model systems we use, trying to understand inter-organ crosstalk, some of the things you cover in your beautiful Twitter reviews. (←Ground Truths link)

I remember recently you highlighted four publications that looked at central nervous system, immune cell crosstalk or central nervous system microbiome crosstalk. It's this sort of long range interaction between organs, between the central nervous system and the immune system and the cancer that I'm hugely interested in because I really think there are vital clues there that will unlock new targets that will enable us to control cancers more effectively if we just understood these complex networks better and had more sophisticated animal model systems to be able to interpret these interactions.
Eric Topol (22:11):
No, it's so important what you're bringing out, the mysteries that still we have to deal with cancer, why patients have all these issues or dying without really knowing what's happened no less, as you say, these new connects that are being discovered at a remarkable pace, as you mentioned, that ground truths. And also, for example, when I spoke with Michelle Monje, she's amazing on the cancer, where hijacking the brain cells and just pretty extraordinary things. Now that gets me to another line of work of yours. I mean there are many, but the issue of evolution of the tumor, and if you could put that in context, a hot area that's helping us elucidate these mechanisms is known as spatial omics or spatial biology. This whole idea of being able to get the spatial temporal progression through single cell sequencing and single cell nuclei, all the single cell omics. So if you could kind of take us through what have we learned with this technique and spatial omics that now has changed or illuminated our understanding of how cancer evolves?
Charles Swanton (23:37):
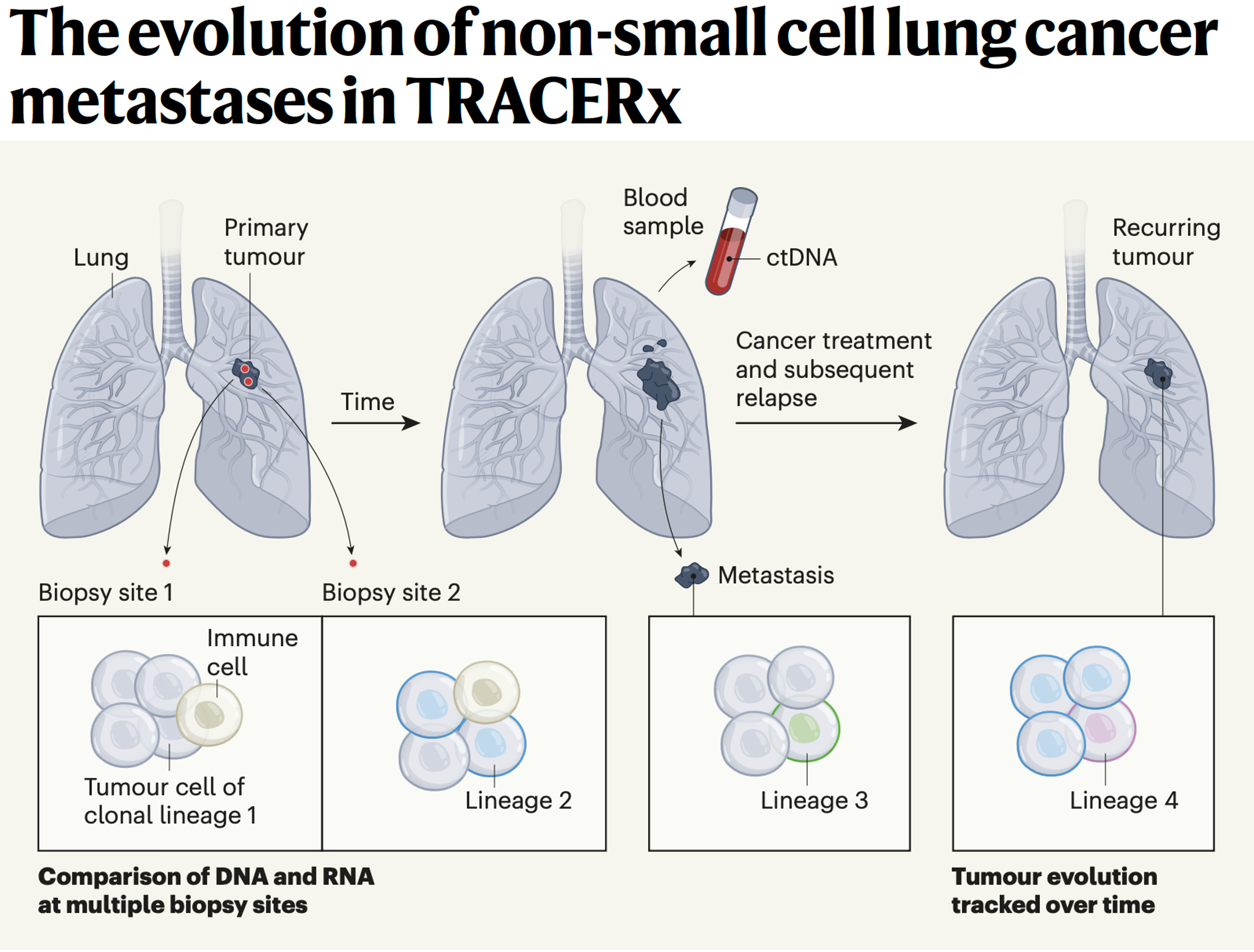
Yeah, great question. Well, I mean I think it helps us sort of rewind a bit and think about evolution in general. Genetic selection brought about by diverse environments and environmental pressures that force evolution, genetic evolution, and speciation down certain evolutionary roots. And I think one can think about cancers in a similar way. They start from a single cell and we can trace the evolutionary paths of cancers by single cell analysis as well as bulk sequencing of spatially separated tumor regions to be able to reconstruct their subclones. And that's taught us to some extent, what are the early events in tumor evolution? What are the biological mechanisms driving branched evolution? How does genome instability begin in tumors? And we found through TRACERx work, whole genome doubling is a major route through to driving chromosome instability along with mutagenic enzymes like APOBEC that drive both mutations and chromosomal instability.
(24:44):
And then that leads to a sort of adaptive radiation in a sense, not dissimilar to I guess the Cambrian explosion of evolutionary opportunity upon which natural selection can act. And that's when you start to see the hallmarks of immune evasion like loss of HLA, the immune recognition molecules that bind the neoantigens or even loss of the neoantigens altogether or mutation of beta 2 microglobulin that allow the tumor cells to now evolve below the radar, so to speak. But you allude to the sort of spatial technologies that allow us to start to interpret the microenvironments as well. And that then tells us what the evolutionary pressures are upon the tumor. And we're learning from those spatial technologies that these environments are incredibly diverse, actually interestingly seem to be converging on one important aspect I'd like to talk to you a little bit more about, which is the myeloid axis, which is these neutrophils, macrophages, et cetera, that seem to be associated with poor outcome and that will perhaps talk about pollution in a minute.
(25:51):
But I think they're creating a sort of chronic inflammatory response that allows these early nascent tumor cells to start to initiate into frankly tumor invasive cells and start to grow. And so, what we're seeing from these spatial technologies in lung cancer is that T-cells, predatory T-cells, force tumors to lose their HLA molecules and what have you to evade the immune system. But for reasons we don't understand, high neutrophil infiltration seems to be associated with poor outcome, poor metastasis free survival. And actually, those same neutrophils we've recently found actually even tracked to the metastasis sites of metastasis. So it's almost like this sort of symbiosis between the myeloid cells and the tumor cells in their biology and growth and progression of the tumor cells.
Eric Topol (26:46):
Yeah, I mean this white cell story, this seems to be getting legs and is relatively new, was this cracked because of the ability to do this type of work to in the past everything was, oh, it's cancer's heterogeneous and now we're getting pinpoint definition of what's going on.
Charles Swanton (27:04):
I think it's certainly contributed, but it's like everything in science, Eric, when you look back, there's evidence in the literature for pretty much everything we've ever discovered. You just need to put the pieces together. And I mean one example would be the neutrophil lymphocyte ratio in the blood as a hallmark of outcome in cancers and to checkpoint inhibitor blockade, maybe this begins to explain it, high neutrophils, immune suppressive environment, high neutrophils, high macrophages, high immune suppression, less benefit from checkpoint inhibitor therapy, whereas you want lymphocyte. So I think there are biomedical medical insights that help inform the biology we do in the lab that have been known for decades or more. And certainly the myeloid M2 axis in macrophages and what have you was known about way before these spatial technologies really came to fruition, I think.
The Impact of Air Pollution
Eric Topol (28:01):
Yeah. Well you touched on this about air pollution and that's another dimension of the work that you and your team have done. As you well know, there was a recent global burden of disease paper in the Lancet, which has now said that air pollution with particulate matter 2.5 less is the leading cause of the burden of disease in the world now.

Charles Swanton (28:32):
What did you think of that, Eric?
Eric Topol (28:34):
I mean, I was blown away. Totally blown away. And this is an era you've really worked on. So can you put it in perspective?
Charles Swanton (28:42):
Yeah. So we got into this because patients of mine, and many of my colleagues would ask the same question, I've never smoked doctor, I'm healthy. I'm in my mid 50s though they're often female and I've got lung cancer. Why is that doctor? I've had a good diet, I exercise, et cetera. And we didn't really have a very good answer for that, and I don't want to pretend for a minute we solved the whole problem. I think hopefully we've contributed to a little bit of understanding of why this may happen. But that aside, we knew that there were risk factors associated with lung cancer that included air pollution, radon exposure, of course, germline genetics, we mustn't forget very important germline variation. And I think there is evidence that all of them are associated with lung cancer risk in different ways. But we wanted to look at air pollution, particularly because there was an awful lot of evidence, several meta-analysis of over half a million individuals showing very convincingly with highly significant results that increasing PM 2.5 micron particulate levels were associated with increased risk of lung cancer.
(29:59):
To put that into perspective, where you are on the west coast of the US, it's relatively unpolluted. You would be talking about maybe five micrograms per meter cubed of PM2.5 in a place like San Diego or Western California, assuming there aren't any forest fires of course. And we estimate that that would translate to about, we think it's about one extra case of never smoking lung cancer per hundred thousand of the population per year per one microgram per meter cube rise in the pollution levels. So if you go to Beijing for example, on a bad day, the air pollution levels could be upwards of a hundred micrograms per meter cubed because there are so many coal fired power stations in China partly. And there I think the risk is considerably higher. And that's certainly what we've seen in the meta-analyses in our limited and relatively crude epidemiological analyses to be the case.
(30:59):
So I think the association was pretty certain, we were very confident from people's prior publications this was important. But of course, association is not causation. So we took a number of animal models and showed that you could promote lung cancer formation in four different oncogene driven lung cancer models. And then the question is how, does air pollution stimulate mutations, which is what I initially thought it would do or something else. It turns out we don't see a significant increase in exogenous like C to A carcinogenic mutations. So that made us put our thinking caps on. And I said to you earlier, often all these discoveries have been made before. Well, Berenblum in 1947, first postulated that actually tumors are initiated through a two-step process, which we now know involves a sort of pre initiated cell with a mutation in that in itself is not sufficient to cause cancer.
(31:58):
But on top of that you need an inflammatory stimulus. So the question was then, well, okay, is inflammation working here? And we found that there was an interleukin-1 beta axis. And what happens is that the macrophages come into the lung on pollution exposure, engulf phagocytose the air pollutants, and we think what's happening is the air pollutants are puncturing membranes in the lung. That's what we think is happening. And interleukin-1 beta preformed IL-1 beta is being released into the extracellular matrix and then stimulating pre-initiated cells stem cells like the AT2 cells with an activating EGFR mutation to form a tumor. But the EGFR mutation alone is not sufficient to form tumors. It's only when you have the interleukin-1 beta and the activated mutation that a tumor can start.
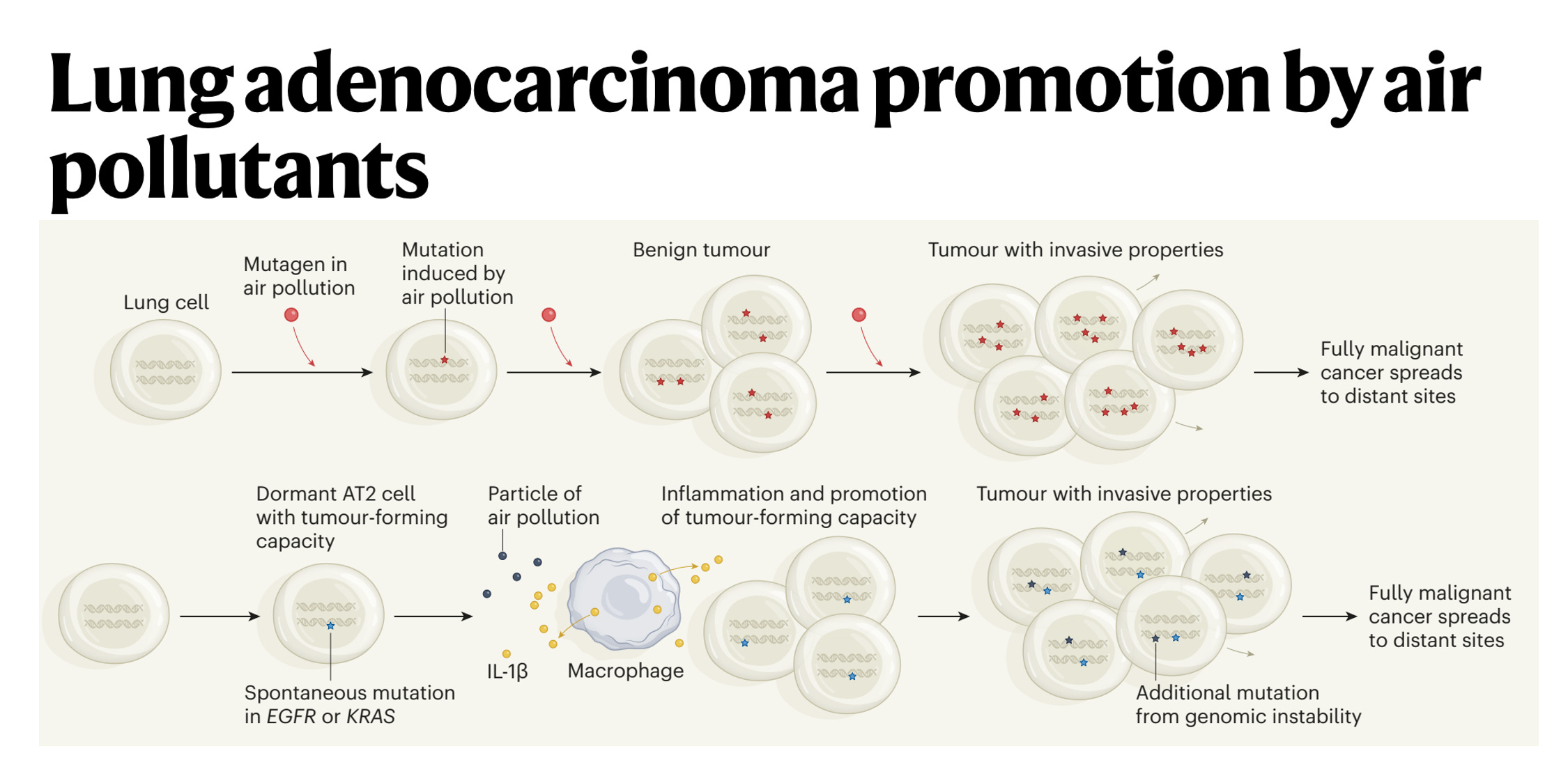
(32:49):
And we found that if we sequence normal lung tissue in a healthy adult 60-year-old adult, we will find about half of biopsies will have an activating KRAS mutation in normal tissue, and about 15% will have an activating mutation in EGFR in histologically normal tissue with nerve and of cancer. In fact, my friend and colleague who's a co-author on the paper, James DeGregori, who you should speak to in Colorado, fascinating evolutionary cancer biologists estimates that in a healthy 60-year-old, there are a hundred billion cells in your body that harbor an oncogenic mutation. So that tells you that at the cellular level, cancer is an incredibly rare event and almost never happens. I mean, our lifetime risk of cancer is perhaps one in two. You covered that beautiful pancreas paper recently where they estimated that there may be 80 to 100 KRAS mutations in a normal adult pancreas, and yet our lifetime risk of pancreas cancer is one in 70. So this tells you that oncogenic mutations are rarely sufficient to drive cancer, so something else must be happening. And in the context of air pollution associated lung cancer, we think that's inflammation driven by these white cells, these myeloid cells, the macrophages.
Cancer Biomarkers
Eric Topol (34:06):
No, it makes a lot of sense. And this, you mentioned the pancreas paper and also what's going in the lung, and it seems like we have this burden of all you need is a tipping point and air pollution seems to qualify, and you seem to be really in the process of icing the mechanism. And like I would've thought it was just mutagenic and it's not so simple, right? But that gets me to this is such an important aspect of cancer, the fact that we harbor these kind of preconditions. And would you think that cancer takes decades to actually manifest most cancers, or do we really have an opportunity here to be able to track whether it's through blood or other biomarkers? Another area you've worked on a lot whereby let's say you could define people at risk for polygenic risk scores or various cancers or genome sequencing for predisposition genes, whatever, and you could monitor in the future over the course of those high-risk people, whether they were starting to manifest microscopic malignancy. Do you have any thoughts about how long it takes for the average person to actually manifest a typical cancer?

Charles Swanton (35:28):
That's a cracking question, and the answer is we've got some clues in various cancers. Peter Campbell would be a good person to speak to. He estimates that some of the earliest steps in renal cancer can occur in adolescence. We've had patients who gave up smoking 30 or so years ago where we can still see the clonal smoking mutations in the trunk of the tumor's evolutionary tree. So the initial footprints of the cancer are made 30 years before the cancer presents. That driver mutation itself may also be a KRAS mutation in a smoking cigarette context, G12C mutation. And those mutations can precede the diagnosis of the disease by decades. So the earliest steps in cancer evolution can occur, we think can precede diagnoses by a long time. So to your point, your question which is, is there an opportunity to intervene? I'm hugely optimistic about this actually, this idea of molecular cancer prevention.
An Anti-Inflammatory Drug Reduces Fatal Cancer and Lung Cancer
(36:41):
How can we use data coming out of various studies in the pancreas, mesothelioma, lung, et cetera to understand the inflammatory responses? I don't think we can do very much about the mutations. The mutations unfortunately are a natural consequence of aging. You and I just sitting here talking for an hour will have accumulated multiple mutations in our bodies over that period, I'm afraid and there's no escaping it. And right now there's not much we can do to eradicate those mutant clones. So if we take that as almost an intractable problem, measuring them is hard enough, eradicating them is even harder. And then we go back to Berenblum in 1947 who said, you need an inflammatory stimulus. Well, could we do something about the inflammation and dampen down the inflammation? And of course, this is why we got so excited about IL-1 beta because of the CANTOS trial, which you may remember in 2017 from Ridker and colleagues showed that anti IL-1 beta used as a mechanism of preventing cardiovascular events was associated with a really impressive dose dependent reduction in new lung cancer primaries.
(37:49):
Really a beautiful example of cancer prevention in action. And that data weren't just a coincidence. The FDA mandated Novartis to collect the solid tumor data and the P-values are 0.001. I mean it's very highly significant dose dependent reduction in lung cancer incidents associated with anti IL-1 beta. So I think that’s really the first clue in my mind that something can be done about this problem. And actually they had five years of follow-up, Eric. So that’s something about that intervening period where you can treat and then over time see a reduction in new lung cancers forming. So I definitely think there’s a window of opportunity here.
Eric Topol (38:31):
Well, what you’re bringing up is fascinating here because this trial, which was a cardiology trial to try to reduce heart attacks, finds a reduction in cancer, and it’s been lost. It’s been buried. I mean, no one’s using this therapy to prevent cancer between ratcheting up the immune system or decreasing inflammation. We have opportunities that we’re not even attempting. Are there any trials that are trying to do this sort of thing?
Charles Swanton (39:02):
So this is the fundamental problem. Nobody wants to invest in prevention because essentially you are dealing with well individuals. It’s like the vaccine challenge all over again. And the problem is you never know who you are benefiting. There’s no economic model for it. So pharma just won’t touch prevention with a barge pole right now. And that’s the problem. There’s no economic model for it. And yet the community, all my academic colleagues are crying out saying, this has got to be possible. This has got to be possible. So CRUK are putting together a group of like-minded individuals to see if we can do something here and we're gradually making progress, but it is tough.
Eric Topol (39:43):
And it's interesting that you bring that up because for GRAIL, one of the multicenter cancer early detection companies, they raised billions of dollars. And in fact, their largest trial is ongoing in the UK, but they haven't really focused on high-risk people. They just took anybody over age 50 or that sort of thing. But that's the only foray to try to reboot how we or make an early microscopic diagnosis of cancer and track people differently. And there's an opportunity there. You've written quite a bit on you and colleagues of the blood markers being able to find a cancer where well before, in fact, I was going to ask you about that is, do you think there's people that are not just having all these mutations every minute, every hour, but that are starting to have the early seeds of cancer, but because their immune system then subsequently kicks in that they basically kind of quash it for that period of time?
Charles Swanton (40:47):
Yeah, I do think that, I mean, the very fact that we see these sort of footprints in the tumor genome of immune evasion tells you that the immune system's having a very profound predatory effect on evolving tumors. So I do think it's very likely that there are tumors occurring that are suppressed by the immune system. There is a clear signature, a signal of negative selection in tumors where clones have been purified during their evolution by the immune system. So I think there's pretty strong evidence for that now. Obviously, it's very difficult to prove something existed when it doesn't now exist, but there absolutely is evidence for that. I think it raises the interesting question of immune system recognizes mutations and our bodies are replete with mutations as we were just discussing. Why is it that we're not just a sort of epithelial lining of autoimmunity with T-cells and immune cells everywhere? And I think what the clever thing about the immune system is it's evolved to target antigens only when they get above a certain burden. Otherwise, I think our epithelial lining, our skin, our guts, all of our tissues will be just full of T-cells eating away our normal clones.
(42:09):
These have to get to a certain size for antigen to be presented at a certain level for the immune system to recognize it. And it's only then that you get the immune predation occurring.
Forever Chemicals and Microplastics
Eric Topol (42:20):
Yeah, well, I mean this is opportunities galore here. I also wanted to extend the air pollution story a bit. Obviously, we talked about particulate matter and there's ozone and nitric NO2, and there's all sorts of other air pollutants, but then there's also in the air and water these forever chemicals PFAS for abbreviation, and they seem to be incriminated like air pollution. Can you comment about that?
Charles Swanton (42:55):
Well, I can comment only insofar as to say I'm worried about the situation. Indeed, I'm worried about microplastics actually, and you actually cover that story as well in the New England Journal, the association of microplastics with plaque rupture and atheroma. And indeed, just as in parenthesis, I wanted to just quickly say we currently think the same mechanisms that are driving lung cancer are probably responsible for atheroma and possibly even neurodegenerative disease. And essentially it all comes down to the macrophages and the microglia becoming clogged up with these pollutants or environmental particulars and releasing chronic inflammatory mediators that ultimately lead to disease. And IL-1 beta being one of those in atheroma and probably IL-6 and TNF in neurodegenerative disease and what have you. But I think this issue that you rightly bring up of what is in our environment and how does it cause pathology is really something that epidemiologists have spent a lot of time focusing on.
(43:56):
But actually in terms of trying to move from association to causation, we've been, I would argue a little bit slow biologically in trying to understand these issues. And I think that is a concern. I mean, to give you an example, Allan Balmain, who works at UCSF quite close to you, published a paper in 2020 showing that 17 out of 20 environmental carcinogens IARC carcinogens class one carcinogens cause tumors in rodent models without driving mutations. So if you take that to a logical conclusion, in my mind, what worries me is that many of the sort of carcinogen assays are based on driving mutagenesis genome instability. But if many carcinogen aren't driving DNA mutagenesis but are still driving cancer, how are they doing it? And do we actually have the right assays to interpret safety of new chemical matter that's being introduced into our environment, these long-lived particles that we're breathing in plastics, pollutants, you name it, until we have the right biological assays, deeming something to be safe I think is tricky.
Eric Topol (45:11):
Absolutely. And I share your concerns on the nanoplastic microplastic story, as you well know, not only have they been seen in arteries that are inflamed and in blood clots and in various tissues, have they been seen so far or even looked for within tumor tissue?
Charles Swanton (45:33):
Good question. I'm not sure they have. I need to check. What I can tell you is we've been doing some experiments in the lab with fluorescent microplastics, 2.5 micron microplastics given inhaled microplastics. We find them in every mouse organ a week after. And these pollutants even get through into the brain through the olfactory bulb we think.
Charles Swanton (45:57):
Permeate every tissue, Eric.
Eric Topol (45:59):
Yeah, no, this is scary because here we are, we have these potentially ingenious ways to prevent cancer in the future, but we're chasing our tails by not doing anything to deal with our environment.
Charles Swanton (46:11):
I think that's right. I totally agree. Yeah.
Eric Topol (46:15):
So I mean, I can talk to you for the rest of the day, but I do want to end up with a topic that we have mutual interest in, which is AI. And also along with that, when you mentioned about aging, I'd like to get your views on these two, how do you see AI fitting into the future of cancer? And then the more general topic is, can we actually at some point modulate the biologic aging process with or without help with from AI? So those are two very dense questions, but maybe you can take us through them.
Charles Swanton (46:57):
How long have we got?
Eric Topol (46:59):
Just however long you have.
A.I. and Cancer
Charles Swanton (47:02):
AI and cancer. Well, AI and medicine actually in general, whether it's biomedical research or medical care, has just infinite potential. And I'm very, very excited about it. I think what excites me about AI is it's almost the infinite possibilities to work across scale. Some of the challenges we raised in the Cell review that you mentioned, tackling, embracing complexity are perfectly suited for an AI problem. Nonlinear data working, for instance in our fields with CT imaging, MRI imaging, clinical outcome data, blood parameters, genomics, transcriptomes and proteomes and trying to relate this all into something that's understandable that relates to risk of disease or potential identification of a new drug target, for example. There are numerous publications that you and others have covered that allude to the incredible possibilities there that are leading to, for instance, the new identification of drug targets. I mean, Eli Van Allen's published some beautiful work here and in the context of prostate cancer with MDM4 and FGF receptor molecules being intimately related to disease biology.
(48:18):
But then it's not just that, not just drug target identification, it's also going all the way through to the clinic through drug discovery. It's how you get these small molecules to interact with oncogenic proteins and to inhibit them. And there are some really spectacular developments going on in, for instance, time resolved cryo-electron microscopy, where in combination with modeling and quantum computing and what have you, you can start to find pockets emerging in mutant proteins, but not the wild type ones that are druggable. And then you can use sort of synthetic AI driven libraries to find small molecules that will be predicted to bind these transiently emerging pockets. So it's almost like AI is primed to help at every stage in scientific investigation from the bench all the way through to the bedside. And there are examples all the way through there in the literature that you and others have covered in the last few years. So I could not be more excited about that.
Eric Topol (49:29):
I couldn't agree with you more. And I think when we get to multimodal AI at the individual level across all their risks for conditions in their future, I hope someday will fulfill that fantasy of primary prevention. And that is getting me to this point that I touched on because I do think they interact to some degree AI and then will we ever be able to have an impact on aging? Most people conflate this because what we've been talking about throughout the hour has been age-related diseases, that is cancer, for example, and cardiovascular and neurodegenerative, which is different than changing aging per se, body wide aging. Do you think we'll ever changed body wide aging?
Charles Swanton (50:18):
Wow, what a question. Well, if you'd asked me 10 years ago, 15 years ago, do you think we'll ever cure melanoma in my lifetime, I'd have said definitely not. And now look where we are. Half of patients with melanoma, advanced melanoma, even with brain metastasis curd with combination checkpoint therapy. So I never say never in biology anymore. It always comes back to bite you and prove you wrong. So I think it's perfectly possible.
Charles Swanton (50:49):
We have ways to slow down the aging process. I guess the question is what will be the consequences of that?
Eric Topol (50:55):
That's what I was going to ask you, because all these things like epigenetic reprogramming and senolytic drugs, and they seem to at least pose some risk for cancer.
Charles Swanton (51:09):
That's the problem. This is an evolutionary phenomenon. It's a sort of biological response to the onslaught of these malignant cells that are potentially occurring every day in our normal tissue. And so, by tackling one problem, do we create another? And I think that's going to be the big challenge over the next 50 years.
Eric Topol (51:31):
Yeah, and I think your point about the multi-decade challenge, because if you can promote healthy aging without any risk of cancer, that would be great. But if the tradeoff is close, it's not going to be very favorable. That seems to be the main liability of modulation aging through many of the, there's many shots on goal here, of course, as you well know. But they do seem to pose that risk in general.
Charles Swanton (51:58):
I think that's right. I think the other thing is, I still find, I don’t know if you agree with me, but it is an immense conundrum. What is the underlying molecular basis for somatic aging, for aging of normal tissues? And it may be multifactorial, it may not be just one answer to that question. And different tissues may age in different ways. I don't know. It's a fascinating area of biology, but I think it really needs to be studied more because as you say, it underpins all of these diseases we've been talking about today, cardiovascular, neurodegeneration, cancer, you name it. We absolutely have to understand this. And actually, the more I work in cancer, the more I feel like actually what I'm working on is aging.
(52:48):
And this is something that James DeGregori and I have discussed a lot. There's an observation that in medicine around patients with alpha-1 antitrypsin deficiency who are at higher risk of lung cancer, but they're also at high risk of COPD, and we know the associations of chronic obstructive pulmonary disease with lung cancer risk. And one of the theories that James had, and I think this is a beautiful idea, actually, is as our tissues age, and COPD is a reflection of aging, to some extent gone wrong. And as our tissues age, they become less good at controlling the expansion of these premalignant clones, harboring, harboring oncogenic mutations in normal tissue. And as those premalignant clones expand, the substrate for evolution also expands. So there's more likely to be a second and third hit genetically. So it may be by disrupting the extracellular matrices through inflammation that triggers COPD through alpha-1 antitrypsin deficiency or smoking, et cetera, you are less effectively controlling these emergent clones that just expand with age, which I think is a fascinating idea actually.
Eric Topol (54:01):
It really is. Well, I want to tell you, Charlie, this has been the most fascinating, exhilarating discussion I've ever had on cancer. I mean, really, I am indebted to you because not just all the work you've done, but your ability to really express it, articulate it in a way that hopefully everyone can understand who's listening or reading the transcript. So we'll keep following what you're doing because you're doing a lot of stuff. I can't thank you enough for joining me today, and you've given me lots of things to think about. I hope the people that are listening or reading feel the same way. I mean, this has been so mind bending in many respects. We're indebted to you.
Charles Swanton (54:49):
Well, we all love reading your Twitter feeds. Keep them coming. It helps us keep a broader view of medicine and biological research, not just cancer, which is why I love it so much.
******************************************
The Ground Truths newsletters and podcasts are all free, open-access, without ads.
Please share this post/podcast with your friends and network if you found it informative
Voluntary paid subscriptions all go to support Scripps Research. Many thanks for that—they greatly helped fund our summer internship programs for 2023 and 2024.
Thanks to my producer Jessica Nguyen and Sinjun Balabanoff tor audio and video support at Scripps Research.
Note: you can select preferences to receive emails about newsletters, podcasts, or all I don’t want to bother you with an email for content that you’re not interested in.









Share this post